中国畜禽种业 ›› 2026, Vol. 22 ›› Issue (5): 11-20.doi: 10.19543/j.cnki.1673-4556.20260427.002
尹啸啸( ), 梁捷特, 楚金雨, 李新云, 马云龙()
), 梁捷特, 楚金雨, 李新云, 马云龙()
Xiaoxiao Yin(), Jiete Liang, Jinyu Chu, Xinyun Li, Yunlong Ma()
摘要:
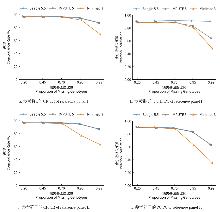
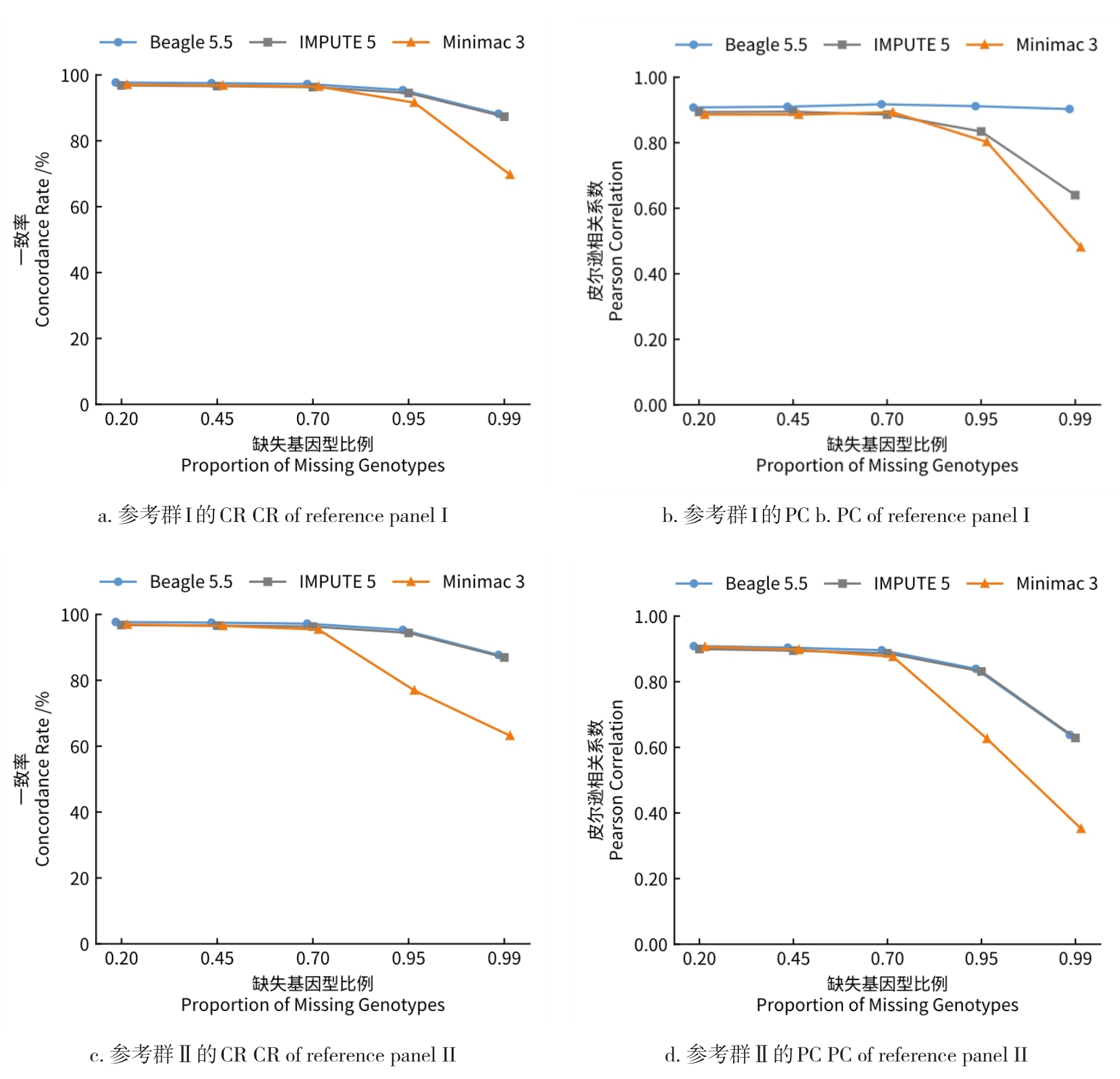
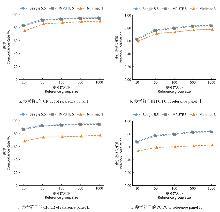
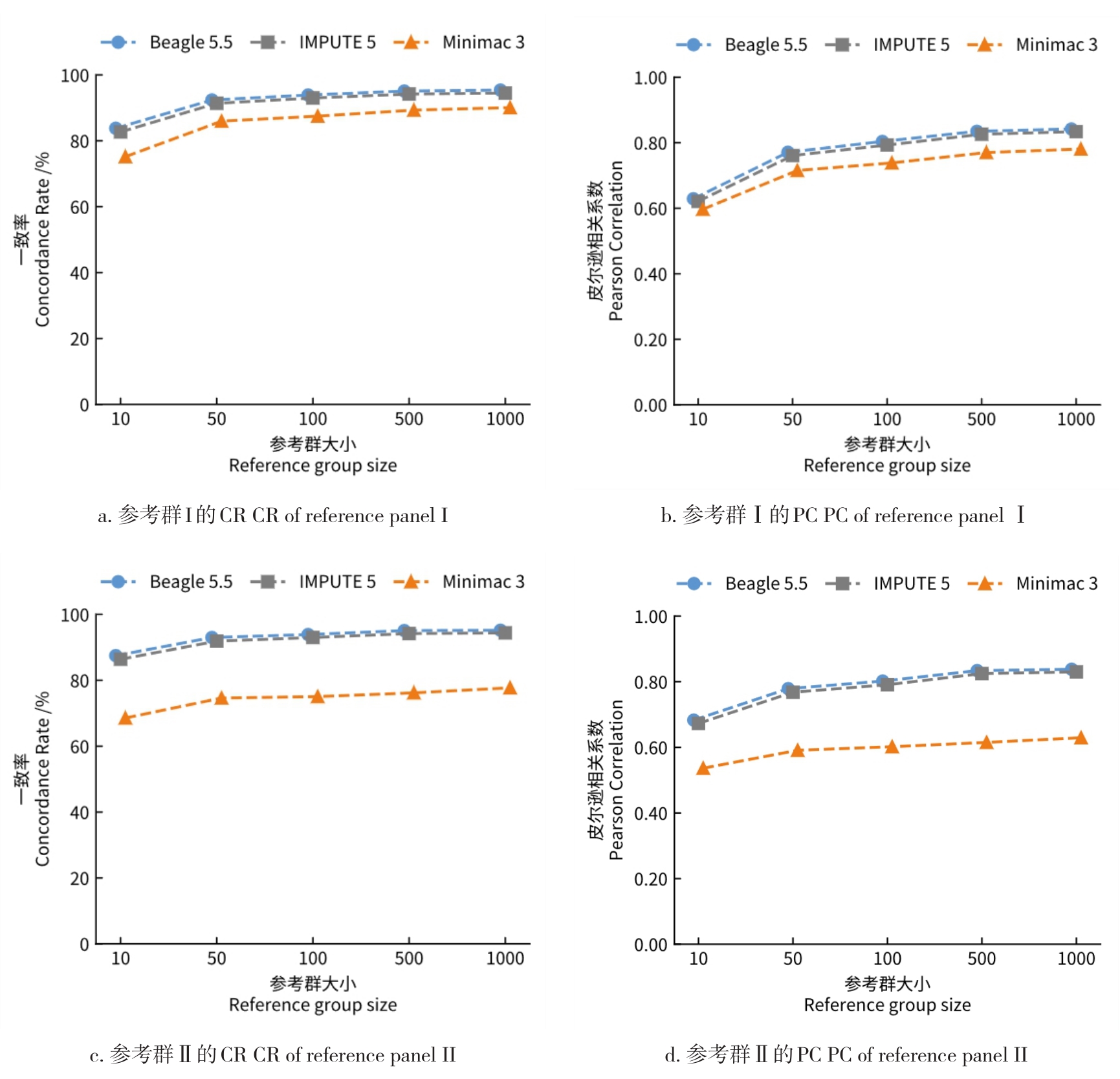
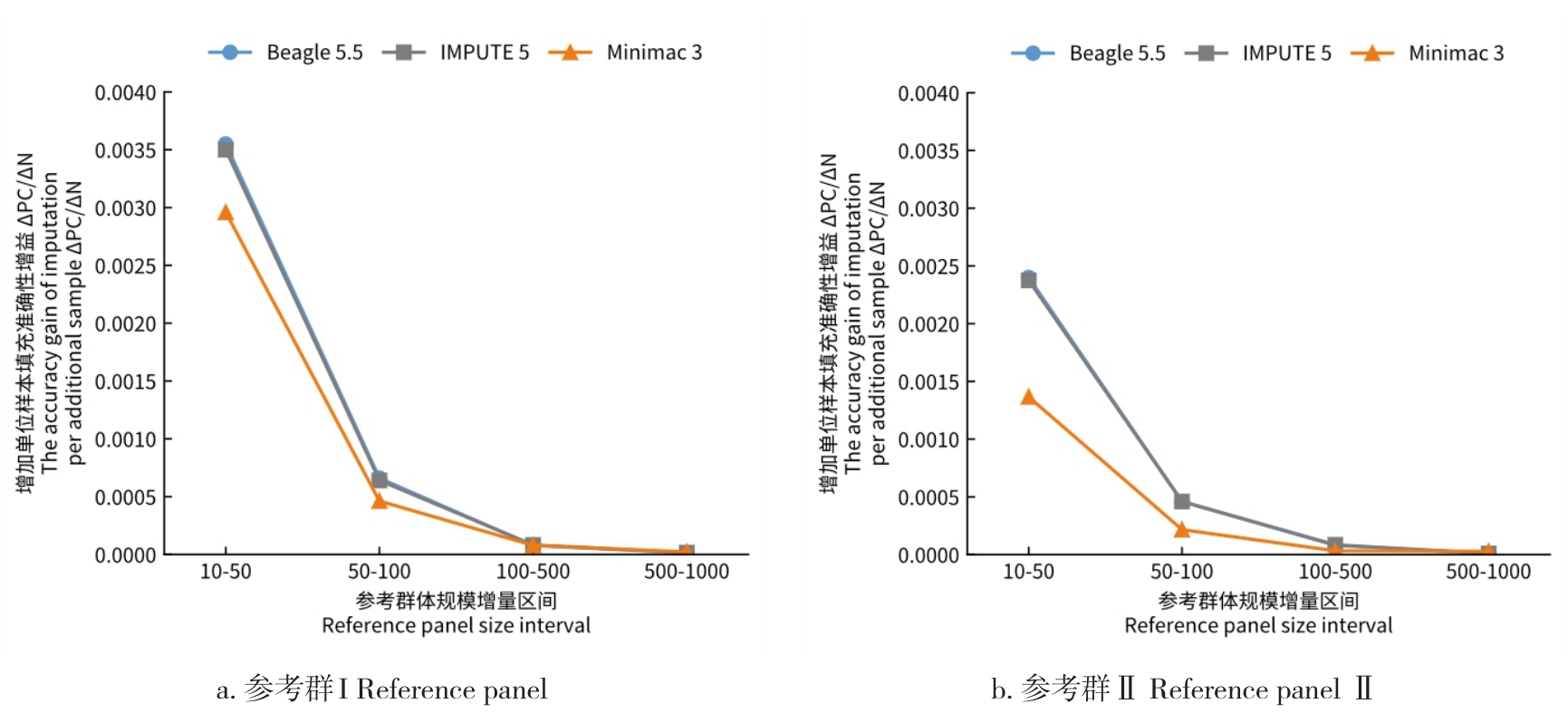
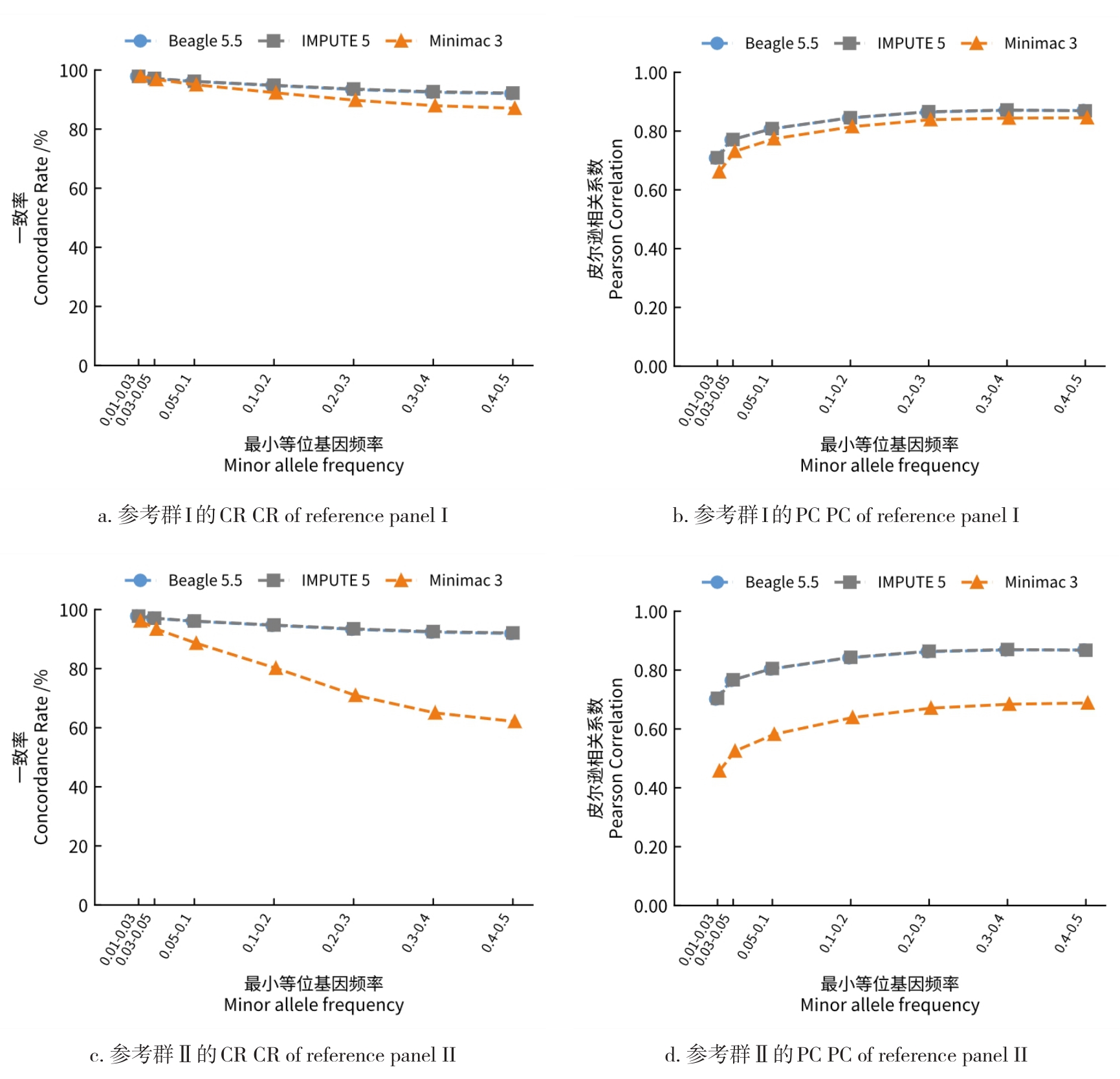
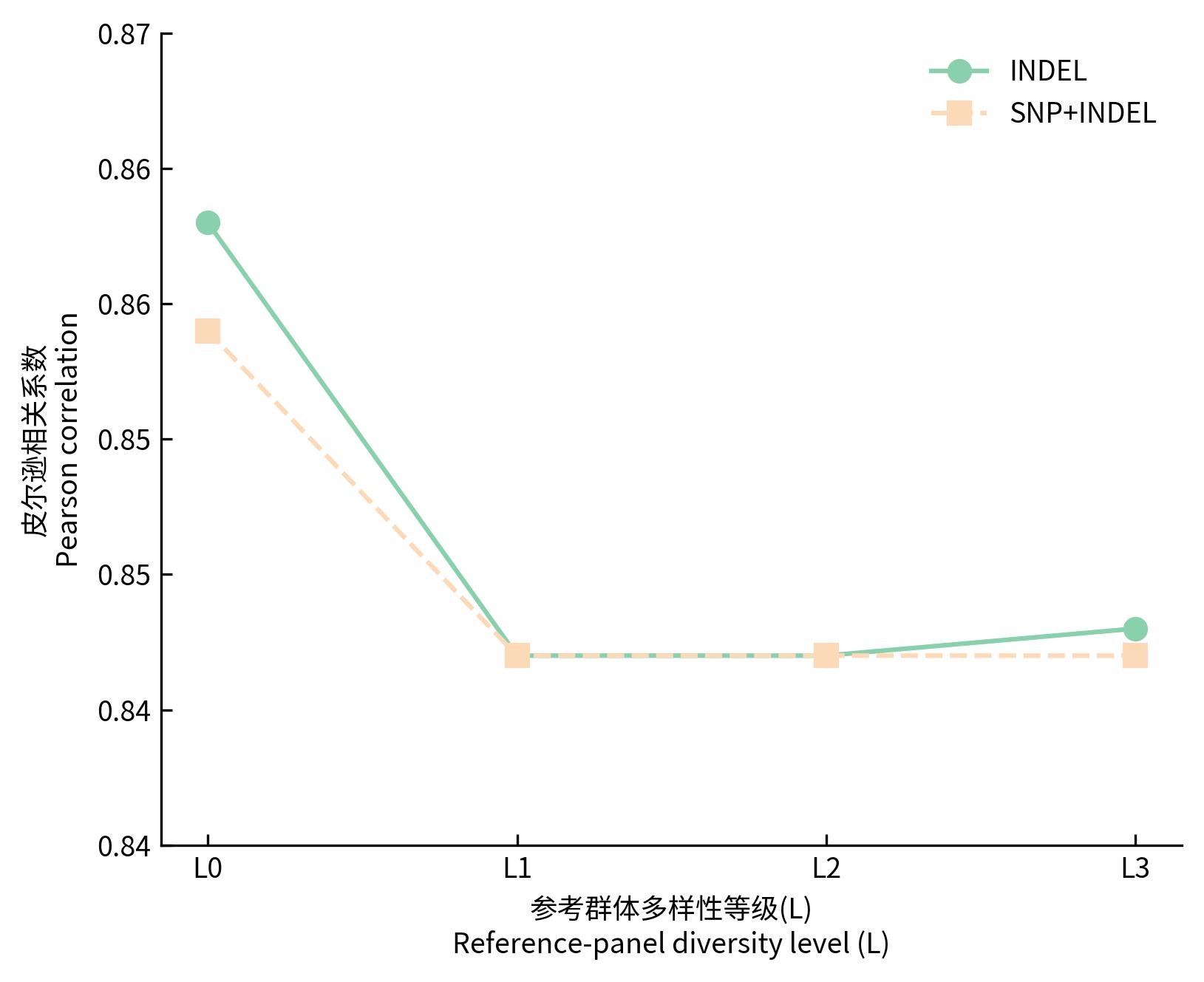
目的 该研究通过对猪插入/缺失进行基因型填充,评估不同因素对填充准确性的影响,并筛选最优填充策略。 方法 基于1119头猪的全基因组测序数据构建参考群I(仅含INDEL基因型)和参考群Ⅱ(含SNP+INDEL基因型)。选取200头大白猪为验证群,对其常染色体上的INDEL基因型按完全随机原则分染色体制造缺失,设置5个缺失比例:20%、45%、70%、95%和99%,以模拟不同标记密度;设置5个参考群规模(10、50、100、500、1000)和7个最小等位基因频率(MAF)区间([0.01,0.03)、[0.03,0.05)、[0.05,0.1)、[0.1,0.2)、[0.2,0.3)、[0.3,0.4)、[0.4,0.5]);在100头大白猪群体的基础上,引入其他猪种,设置了4个等级的参考群多样性(L0~L3);对比了Beagle 5.5、IMPUTE 5和Minimac 3三种软件的INDEL填充准确性即一致率(Concordance rate,CR)和皮尔逊相关性(Pearson correlation,PC)。 结果 在各试验条件下,参考群Ⅰ的填充准确性优于参考群Ⅱ,二者平均PC值分别为0.797和0.760。在参考群Ⅰ中,Beagle 5.5和IMPUTE 5的PC值分别由20%缺失时的0.898和0.900降至99%缺失时的0.641和0.640,而Minimac 3在99%缺失时仅为0.481;Beagle 5.5的PC值由参考群规模为10头时的0.619升至100头时的0.794,增至1000头时仅升至0.832;当MAF由0.01~0.03增至0.4~0.5时,Beagle 5.5的PC值由0.571升至0.837;L0级PC值为0.863,高于L1、L2、L3级的0.847、0.847、0.848。 结论 综上可见,参考群中SNP的引入会降低INDEL填充的准确性,随着标记密度的降低,填充准确性也随之降低,Beagle 5.5和IMPUTE 5均适用于猪INDEL填充,而Minimac 3在极低标记密度下表现较差;填充准确性随参考群规模增大而提升,参考群规模增至约100头后,新增样本带来的准确性增益明显减弱;与验证群遗传背景最一致的L0级准确性最高。在猪INDEL基因型填充中,建议使用较高的标记密度数据,参考群规模建议选择100头左右。推荐使用MAF>0.05作为INDEL基因型填充后的质控标准。当参考群为多品种混合时,尽可能选择与验证群遗传背景相似的群体作为参考群。选择合适的软件并结合其运行效率与填充准确性,仍是确保研究结果可靠性的关键。本研究可为猪INDEL基因型填充策略的优化提供参考,并为基于INDEL变异开展复杂性状遗传解析与育种应用研究提供方法学依据。
中图分类号:
| [1] | MARCHINI J, HOWIE B, MYERS S, et al. A new multipoint method for genome-wide association studies by imputation of genotypes[J]. Nature Genetics, 2007, 39(7): 906-913. |
| [2] | HOWIE B N, DONNELLY P, MARCHINI J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies[J]. PLoS Genetics, 2009, 5(6): e1000529. |
| [3] | BROWNING B L, ZHOU Y, BROWNING S R. A one-penny imputed genome from next-generation reference panels[J]. A-merican Journal of human genetics, 2018, 103(3): 338-348. |
| [4] | RUBINACCI S, DELANEAU O, MARCHINI J. Genotype imputation using the positional burrows wheeler transform[J]. PLoS Genetics, 2020, 16(11): e1009049. |
| [5] | DAS S, FORER L, SCHÖNHERR S, et al. Next-generation genotype imputation service and methods[J]. Nature Genetics, 2016, 48(10): 1284-1287. |
| [6] | NGUYEN T V, BOLORMAA S, REICH C M, et al. Empirical versus estimated accuracy of imputation: optimising filtering thresholds for sequence imputation[J]. Genetics Selection Ev-olution, 2024, 56(1): 72. |
| [7] | LEE D, KIM Y, CHUNG Y, et al. Accuracy of genotype imputation based on reference population size and marker density in Hanwoo cattle[J]. Journal of Animal Science and Technology, 2021, 63(6): 1232-1246. |
| [8] | COSTA HERMISDORFF I DA, COSTA R B, DE ALBUQUERQUE L G, et al. Investigating the accuracy of imputing autosomal variants in Nellore cattle using the ARS-UCD1.2 assembly of the bovine genome[J]. BMC Genomics, 2020, 21(1): 772. |
| [9] | ZHANG K L, PENG X, ZHANG S X, et al. A comprehensive evaluation of factors affecting the accuracy of pig genotype imputation using a single or multi-breed reference population[J]. Journal of Integrative Agriculture, 2022, 21(2): 486-495. |
| [10] | MULLANEY J M, MILLS R E, PITTARD W S, et al. Small insertions and deletions (INDELs) in human genomes[J]. Human Molecular Genetics, 2010, 19(R2): R131-R136. |
| [11] | XU J Y, FU Y H, HU Y, et al. Whole genome variants across 57 pig breeds enable comprehensive identification of genetic signatures that underlie breed features[J]. Journal of Animal Science and Biotechnology, 2020, 11(1): 115. |
| [12] | ROY M E, MANOJ M, ROJAN P M, et al. Identification of genetic variants by whole genome sequencing in Ankamali pigs of Kerala [J]. Journal of Veterinary and Animal Sciences, 2023, 54(2): 524-531. |
| [13] | FANG H, WU Y Y, NARZISI G, et al. Reducing INDEL calling errors in whole genome and exome sequencing data[J]. Genome Medicine, 2014, 6(10):89. |
| [14] | PURCELL S, NEALE B, TODD-BROWN K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses[J]. American Journal of Human Genetics, 2007, 81(3): 559-575. |
| [15] | CHANG C C, CHOW C C, TELLIER L C, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets[J]. GigaScience, 2015, 4: 7. |
| [16] | HOFMEISTER R J, RIBEIRO D M, RUBINACCI S, et al. Accurate rare variant phasing of whole-genome and whole-exome sequencing data in the UK Biobank[J]. Nature Genetics, 2023, 55(7): 1243-1249. |
| [17] | DING R R, SAVEGNAGO R, LIU J D, et al. The SWine IMputation (SWIM) haplotype reference panel enables nucleoti-de resolution genetic mapping in pigs[J]. Communications Biol-ogy, 2023, 6: 577. |
| [18] | ZHANG K L, LIANG J T, FU Y H, et al. AGIDB: a versatile database for genotype imputation and variant decoding across species[J]. Nucleic Acids Research, 2024, 52(D1): D835-D849. |
| [19] | ULLAH E, MALL R, ABBAS M M, et al. Comparison and assessment of family- and population-based genotype imputat-ion methods in large pedigrees[J]. Genome Research, 2019, 29(1): 125-134. |
| [20] | TONG X K, CHEN D, HU J C, et al. Accurate haplotype construction and detection of selection signatures enabled by high quality pig genome sequences[J]. Nature Communications, 2023, 14: 5126. |
| [21] | WANG Q Y, ZHANG Z Y, YE X W, et al. An updated Pig Haplotype Reference Panel (PHARP 4.0) comprising 13, 298 haplotypes[J]. Communications Biology, 2025, 8: 1625. |
| [22] | CAI Z X, SARUP P, OSTERSEN T, et al. Genomic diversity revealed by whole-genome sequencing in three Danish commercial pig breeds[J]. Journal of Animal Science, 2020, 98(7):skaa229. |
| [23] | CHEN L F, YANG S P, ARAYA S, et al. Genotype imputation for soybean nested association mapping population to improve precision of QTL detection[J]. Theoretical and Applied Genetics, 2022, 135(5): 1797-1810. |
| [24] | HICKEY J M, CROSSA J, BABU R, et al. Factors affecting the accuracy of genotype imputation in populations from several maize breeding programs[J]. Crop Science, 2012, 52(2): 654-663. |
| [25] | RAMNARINE S, ZHANG J, CHEN L S, et al. When does choice of accuracy measure alter imputation accuracy assessments[J]. PLoS One, 2015, 10(10): e0137601. |
| [26] | CAHOON J L, RUI X Y, TANG E, et al. Imputation accuracy across global human populations[J]. The American Journal Of Human Genetics, 2024, 111(5): 979-989. |
| [27] | LU J T, WANG Y, GIBBS R A, et al. Characterizing linkage disequilibrium and evaluating the imputation power of human genomic insertion-deletion polymorphisms[J]. Genome Biology, 2012, 13(2): R15. |
| [28] | STAHL K, GOLA D, KÖNIG I R. Assessment of imputation quality: comparison of phasing and imputation algorithms in real data[J]. Frontiers in Genetics, 2021, 12: 724037. |
| [29] | DE MARINO A, MAHMOUD A A, BOSE M, et al. A comparative analysis of current phasing and imputation software[J]. PLoS One, 2022, 17(10): e0260177. |
| [30] | WANG X Q, WANG L G, SHI L Y, et al. Imputation strategies for low-coverage whole-genome sequencing data and their effects on genomic prediction and genome-wide association studies in pigs[J]. Animal, 2024, 18(9): 101258. |
| [31] | DENG T Y, ZHANG P F, GARRICK D, et al. Comparison of genotype imputation for SNP array and low-coverage whole-genome sequencing data[J]. Frontiers in Genetics, 2022, 12: 704118. |
| [1] | 柯尝玲, 曹奎, 刘敬, 付戴波, 曾思静, 张健, 余公修, 胡耀, 熊雄, 徐仕明, 周泉勇. 杜洛克与赣南藏香猪杂交组合性能测定[J]. 中国畜禽种业, 2026, 22(5): 125-130. |
| [2] | 曹翠萍. 丹麦生猪高PSY成因分析及启示[J]. 中国畜禽种业, 2026, 22(5): 7-10. |
| [3] | 刘志国, 黄雷, 文一龙, 牟玉莲. 猪胚胎冷冻保存技术研究进展[J]. 中国畜禽种业, 2026, 22(5): 88-95. |
| [4] | 程中平. 阳泉市非洲猪瘟生物安全综合防控技术的应用[J]. 中国畜禽种业, 2023, 19(9): 132-136. |
| [5] | 季佩东. 基于非瘟防控的我国地方猪种转群方案设计初探——以淮猪为例[J]. 中国畜禽种业, 2023, 19(8): 49-52. |
| [6] | 方晓敏, 顾岳清, 黄媛, 李顺. 二花脸猪种质特性发展现状及保种建议[J]. 中国畜禽种业, 2023, 19(8): 53-57. |
| [7] | 白红杰. 农业强省背景下种企信息化管理和品牌建设—以河南农科种猪科技有限公司为例[J]. 中国畜禽种业, 2023, 19(8): 106-110. |
| [8] | 张维秋, 崔春祥, 傅嘉堃. 烟台黑猪产业现状与发展思路[J]. 中国畜禽种业, 2023, 19(8): 111-115. |
| [9] | 张午霞, 孙涛. 沙乌头猪保种选育技术及保种成效探析[J]. 中国畜禽种业, 2023, 19(6): 89-94. |
| [10] | 张万强, 周彪, 林海. 宁乡花猪如何在行业新常态下发展壮大[J]. 中国畜禽种业, 2023, 19(6): 100-104. |
| [11] | 吴雨, 周迪, 陈琨, 蒋桂荣, 杨蓉, 王燕, 敖叶, 方华, 王舍. 种公猪精液品质候选基因的研究进展[J]. 中国畜禽种业, 2023, 19(6): 105-112. |
| [12] | 孟荣. 贵州省独山县长白猪主要疫病流行病学调查[J]. 中国畜禽种业, 2023, 19(6): 155-158. |
| [13] | 张海筠. 辽宁黑猪种猪综合选择指数的计算[J]. 中国畜禽种业, 2023, 19(5): 36-40. |
| [14] | 唐骏, 占松鹤, 吴惠娟, 汪美莲. 安徽生猪种业创新工作思考与策略[J]. 中国畜禽种业, 2023, 19(5): 45-48. |
| [15] | 占松鹤, 涂小璐, 席海龙, 唐骏, 倪泽兰. 安徽省生猪种业现状与思考[J]. 中国畜禽种业, 2023, 19(5): 49-52. |
|
||